now that I read more of it, a lot of what you say is complete and utter bullcrap holy sh*t. no, they do NOT need two phases to quality for conditional release, I literally talked to a dude yesterday who got a treatment that is under such a scheme right now and they have only done a 12 month 20 person trial. you make so much sh*t up, it's literally in the bill
https://www.caladrius.com/wp-content/uploads/2018/05/Sietsema-et-al.-2018-RAPS.pdf
read this and then look at the graphic on page 14, that should disprove this nonsense. only one trial is required in which both things are tested at once, this is unique to regenerative stem cell treatments but think about it in the case how tsuji, how deranged would it be to make two trials after one another one where you see if the hair grows and another one in which you check efficiency. obviously that nonsense and not required and if you actually read this bill you'd know that. but you haven't
Well first of all I didn’t make anything up, I provided three sources about articles on these new regenerative therapy regulations in Japan as of 2014, and I quoted them. So let us get more scrupulous:
- Page 2 of your source where it explains the logic behind separating pharmaceuticals from regenerative therapy’s, treating the regulatory process different, hopefully hastening advanced cellular therapies, etc.: ...products are approved through the Clinical Trial Notification (CTN) system, as are pharmaceuticals and medical devices. Consultations, required for regenerative medicine products prior to submission of a CTN, are also managed in a similar fashion as those for pharmaceutical products and medical devices, although the consultation may be more detailed for regenerative medicine products.
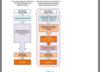
It is possible for regenerative medicine products comprised of heterogeneous components to receive conditional approval with a limited clinical data set, as long as the data demonstrate a safety profile of the new product consistent with the severity of the disease or condition being treated along with efficacy data predicting probable patient benefit. Probable patient benefit may be demonstrated with data sets from exploratory clinical trials and may be based on predictive surrogate endpoints. In practice, a conditional approval with a limited data set will be granted after consultation with the PMDA to discuss the design of the trial and the data set to be considered for getting conditional approval. More than one consultation from early stages of development will likely be needed to achieve agreement on the study design. Once data generation is complete, a Japan New Drug Application (JNDA) is prepared and submitted and subjected to a review process similar a review for pharmaceutical products (Figure 1).5 The JNDA is first reviewed by the PMDA and a report is written which may recommend for or against approval of the product. The report may set out conditions for
I think your analysis of this test is a bit ambiguous as the text itself Is. “ receive conditional approval with limited clinical set, as long as the data demonstrates a safety profile of the new product consistent with the severity of the disease or condition being treated along with efficacy data predicting probability patient benefit.“ “ in practice, a conditional approval with a limited data set will be granted after consultation with the PMDA to discuss the design of the trial and the data set to be considered for getting conditional approval.
More than one consultation from early stages of development will likely be needed to achieve agreement on the study design. “
So it seems the focus is conditional approval, as we have discussed above the seven year Period, clearly they emphasize limited clinical data, but are emphatic about the clinical dabta demonstrating a safety profile, at bare minimum this requires a small size phase 0 and maybe even phase 1, and even a phase 2 ( all three of these phases are primarily for safety a phase 2 is when efficacy begins to actually become a priority, naturally any kind of beneficial outcome no matter what phase will be recorded but a phase 0 end phase 1 are focussed on gathering safety data, phase 2 is for safety and efficacy ) not to mention the animal research before ( which we know for this specific work is concluded, unless their results do not replicate within humans.) The safety data needs to be consistent with the severity of the disease/ condition Sakigake designation is for terminal conditions as described in your source and one of mine as well, within this designation very little clinical data is required for obvious reasons. With a very superficial, cosmetic condition safety is naturally going to be of far more concerned, ergo needing more safety data. Also it clearly states the efficacy data predicting the probability of benefit is taken into account as well so unless you have stellar results in a very limited trial with not any adverse affects in human patients and a very high probability of benefit by this description, you’re going to need to access a larger population size, double blinded placebo in order to get the safety and/or efficacy data a.k.a. phase 2.
-
Once data generation is complete, a Japan New Drug Application (JNDA) is prepared and submitted and subjected to a review process similar a review for pharmaceutical products (Figure 1).5 The JNDA is first reviewed by the PMDA and a report is written which may recommend for or against approval of the product. The report may set out conditions for approval. The report is then submitted to the MHLW for consideration. An advisory committee may be engaged to assist MHLW in its deliberations. Aconditional approval will be time-bounded and include requirements for further studying the safety and efficacy of the regenerative medical product during the conditional approval period. The conditional approval period may be as long as seven years or fewer, as determined at the discretion of the Japanese regulators. At the end of the conditional approval period, a re-application must be submitted to continue marketing the regenerative medical product and the PMDA/MHLW review and decision process will be repeated. The regenerative medical product's marketing authorization may be revoked if the safety and efficacy of the product has not been corroborated.
So the initial review process is similar to pharmaceuticals, and then it has to undergo its own share of bureaucratic stipulation going through two different regulatory boards, who can derail the entire process by requiring an advisory commission. Who are both fully capable of outright denying the approval, or designing strict stipulations. Either way according to the text just as my sources said as well during conditional approval further studying the safety and efficacy of the regenerative product is obligated to continue, ergo people are now able to buy the product within the Japanese market but clinical trials and data gathering do not stop, nor do their expenses.
We have analyzed your source now let’s take a look at what I provided from a different source summarizing this legislation:
- New regulations accelerating the approval of regenerative therapeutics in Japan took effect November 25, 2014. The significance of these regulations is that they allow companies to receive conditional marketing approval and commercialize regenerative medicine products while clinical trials continue through the later stages.
- Similarly, the PMD Act introduces a specific regulatory framework for regenerative medicine products. Under the PMD Act, conditional and time-limited marketing approval can be given to a regenerative medicine product after exploratory clinical trials have demonstrated probable benefit and proven safety. According to these new laws, once a company has demonstrated safety and basic efficiency data in humans and has the cell product manufactured to the standards described within the Pharmaceutical and Medical Devices (PMD) Act, the cell therapy can be given conditional approval for up to seven years. This allows for commercial use with data reporting requirements and potential for national insurance coverage.
- The intent of the laws is to accelerate the commercialization of cell therapeutics within Japan by allowing companies to benefit from conditional marketing authorization. Therefore, cell therapies that show safety and probable efficacy during Phase I and Phase II trials can get conditional approval for up to seven years, during which time:
1) Larger-scale, later-stage clinical trials are performed
2)Revenue from the cell therapy is pursued within the Japanese market
During the seven-year conditional approval period, companies must continue to submit clinical trial data to Japan’s Pharmaceuticals and Medical Devices Agency (PMDA), and subsequently apply for final marketing approval or withdraw the product within seven years.
https://bioinformant.com/japan-accelerated-approvals-of-cell-therapies/
Literally summarizes everything in the source you provided, it literally reiterates everything. At one point it states how cell therapies will show safety and probable efficacy data through phase 1 and phase 2 trials, as Long as it is not within the Sakigake designation looking to treat terminal conditions with no affective treatment currently, this is a fair statement, it is almost guaranteed a cellular-based product outside of that designation Will require a phase 1 human trial, depending on how well that trial goes they may or may not have to gather more data potentially requiring more patients and different methodology: double blinded-placebo for example, a.k.a. a phase 2… This will be at the discretion of the PMDA and MHLW ( also potential he at the discretion of their advisory committee if they request). This is a multifaceted decision making process, analyzing each individual condition, regenerative product, team of researchers, pharmaceutical company backing, and the data that is gathered and presented of course. If you think that both regulatory organizations will never ask for two phases of clinical data and analysis you are delusional, regenerative research especially in regards to genetic therapies are a new frontier of medicine..